
O setor agrícola conta com uma nova ferramenta biotecnológica para combater pragas agrícolas, que causam grandes perdas às lavouras no mundo todo. As multinacionais norte-americanas Monsanto, recém-adquirida pela alemã Bayer, e DowDuPont obtiveram no ano passado aprovação para uso comercial nos Estados Unidos de uma semente transgênica feita com a técnica de silenciamento gênico por RNA (ácido ribonucleico) de interferência, ou simplesmente RNAi. Com efeito inseticida, ela foi criada para controlar a larva-alfinete americana (Diabrotica virgifera), fase larval de um besouro que é a principal ameaça às plantações de milho naquele país. É a primeira vez que moléculas de RNAi são usadas no combate a pragas do campo.

No Brasil, a empresa Tropical Melhoramento & Genética (TMG), com sede no Paraná e especializada em melhoramento genético de soja e algodão, espera dispor ainda este ano de uma molécula de RNAi para uso contra o percevejo-da-soja (Euschistus heros), importante praga da oleaginosa. O desenvolvimento da tecnologia está sendo feito por pesquisadores da Universidade Estadual de Campinas (Unicamp). A Empresa Brasileira de Pesquisa Agropecuária (Embrapa) também já criou um feijão transgênico com a técnica de RNAi, mas para controle de um vírus, conhecido como mosaico-dourado, que ataca esse plantio. Embora já aprovado desde 2011 pela Comissão Técnica Nacional de Biossegurança (CTNBio), do Ministério da Ciência, Tecnologia, Inovações e Comunicações (MCTIC), o produto ainda não foi comercializado.
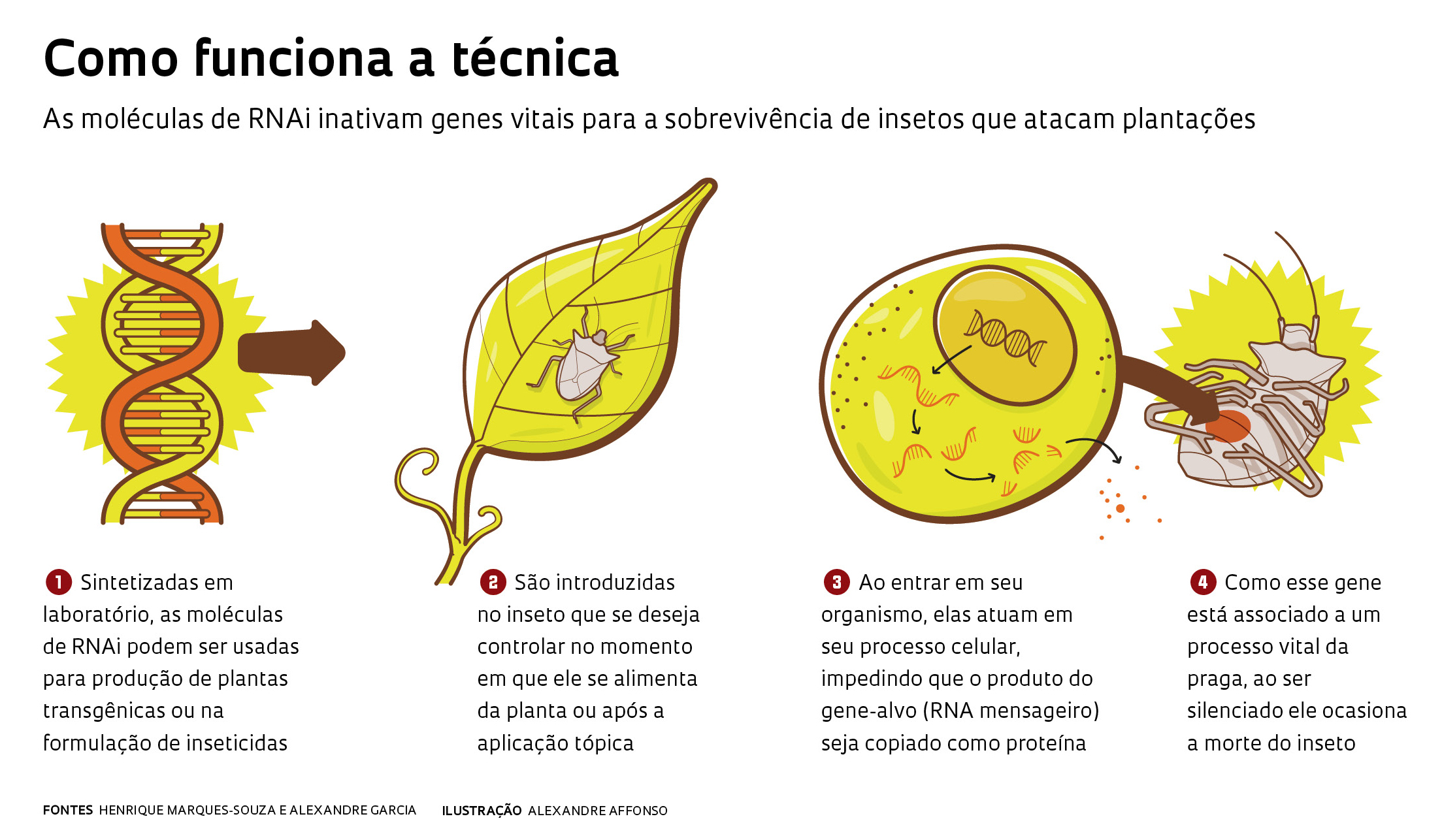
Criadas em laboratório, as moléculas de RNAi neutralizam a ação de um gene-alvo em um organismo qualquer – daí a expressão silenciamento gênico. Seu mecanismo de ação é similar ao que ocorre naturalmente em todos os seres vivos ao lutar contra ataques virais. Quando isso ocorre, as defesas do organismo tentam neutralizar a ação do invasor, silenciando seus genes. De modo análogo, a tecnologia de RNAi faz com que a célula ataque o produto de seu próprio gene, como se estivesse destruindo o material genético viral, num processo similar ao de uma resposta autoimune. No uso agrícola, o RNAi é programado para inativar genes específicos de pragas e patógenos associados a processos essenciais à sua sobrevivência.
A descoberta de que o processo de interferência por RNA poderia ser usado a favor do organismo humano rendeu aos pesquisadores Andrew Fire e Craig Mello, do Instituto Carnegie, de Washington, o prêmio Nobel de Medicina de 2006. O achado foi promissor porque revelou uma nova forma de assumir o comando celular, abrindo um amplo leque de aplicações. Desde então, a ferramenta é aplicada na pesquisa básica para estudo da função gênica e, na área da saúde, é vista como uma promessa para o tratamento de doenças genéticas (ver Pesquisa FAPESP nº 133).
Para entender como essa nova tecnologia funciona é preciso lembrar de processos da biologia celular, especificamente o mecanismo de expressão genética. Genes são segmentos da molécula de DNA que controlam as funções metabólicas das células, principalmente por meio da produção de proteínas. “Nossa informação genética está toda armazenada em nosso DNA. Para dar vida e uso a essa informação, o DNA deve, inicialmente, ser transcrito em RNA. Em seguida, esse RNA mensageiro é traduzido para proteínas. Esse é o processo básico de expressão gênica”, explica o biólogo Alexandre Garcia, gestor de pesquisa da TMG.

Como o nome deixa claro, a molécula sintética de RNAi interfere no processo de tradução do RNA mensageiro em proteína, fragmentando-o. Ela intercepta e destrói as informações celulares conduzidas pelo RNA dentro da célula antes que sejam processadas e originem proteínas. Dessa forma, o processo de expressão gênica que dependia dos dados contidos naquele RNA não irá mais ocorrer.
Milho pioneiro
O milho transgênico lançado pela Monsanto e DowDuPont foi batizado internamente de DvSnf7 e seu nome comercial é SmartStax. A sopa de letras é formada pelas iniciais da lagarta Diabrotica virgifera e o nome do gene-alvo, Snf7. Quando o inseto se alimenta da planta, a molécula de RNAi entra em seu organismo e silencia o gene Snf7, impedindo a produção de uma proteína que é vital para o funcionamento de tecidos da larva-alfinete americana. O resultado é a morte do inseto, conhecido como “o besouro de US$ 1 bilhão” por causa do enorme prejuízo causado aos agricultores norte-americanos. A química brasileira Renata Bolognesi integrou a equipe da Monsanto que criou o milho transgênico.
“A aplicação da tecnologia de RNAi na agricultura é bastante promissora”, destaca o engenheiro-agrônomo Henrique Marques-Souza, professor do Instituto de Biologia (IB) da Unicamp e líder de um grupo de pesquisa nacional sobre silenciamento gênico. Ele se especializou no tema durante o doutorado na Universidade de Colônia, na Alemanha, concluído em 2007, e o pós-doutorado na Universidade da Califórnia, em Berkeley, Estados Unidos, realizado entre 2008 e 2010. De volta ao Brasil, passou a lecionar na Unicamp, onde criou o Brazilian Laboratory on Silencing Technologies (Blast) e derivou seus estudos sobre silenciamento gênico para a agricultura.
Em 2012, Marques-Souza e o engenheiro-agrônomo Antonio Vargas de Oliveira Figueira, do Centro de Energia Nuclear na Agricultura da Universidade
de São Paulo (Cena-USP), montaram um banco de sequências da traça-do-tomateiro (Tuta absoluta) e realizaram ensaios de silenciamento gênico para diversos genes do inseto. “Esse trabalho deu visibilidade ao nosso grupo. Hoje, podemos desenvolver a tecnologia de RNAi para virtualmente qualquer praga de qualquer cultura”, afirma o pesquisador da Unicamp. No ano passado, a técnica foi licenciada para a TMG pela Inova, a agência de inovação da Unicamp.
O maior desafio para a criação de um inseticida com RNAi para aplicação tópica é a reduzida estabilidade da molécula no ambiente
O primeiro produto a ser desenvolvido com a ferramenta pela empresa será destinado a controlar o percevejo-da-soja, uma das maiores ameaças à sojicultora nacional. “Esperamos ter definido até o fim do ano as melhores moléculas de RNA. Todo o trabalho será feito em caráter experimental e por meio de estudos controlados”, conta Garcia, da TMG, destacando que o caminho para essas moléculas se tornarem um produto comercial é longo e depende tanto da tecnologia usada para promover sua aplicação como do cenário de aprovações legais.
Segundo Marques-Souza, há duas formas de fazer com que as moléculas de RNAi entrem em contato com as pragas: pela criação de plantas transgênicas, como a Monsanto e a DowDuPont procederam com o milho DvSnf7, ou pelo desenvolvimento de produtos inseticidas. Essa é a escolha da TMG, que planeja criar um produto para aplicação tópica, como, por exemplo, um pulverizador. “Além de diminuir o tempo gasto para a produção de uma planta transgênica, o RNAi tópico pode ser considerado um produto natural, pois se trata de uma molécula biológica que não causa alterações genéticas na cultura de interesse ou no organismo desejado”, explica Marques-Souza.
A criação de um inseticida com RNAi, no entanto, não é trivial, e alguns obstáculos precisam ser superados. “Essas moléculas se degradam rapidamente no ambiente, o que dificulta a criação de um produto para aplicação na lavoura. Por isso, já existem investigações que buscam empregar recursos da nanotecnologia para proteger a molécula de RNAi [ver Pesquisa FAPESP nº 266] e elevar sua estabilidade”, destaca o biólogo Fernando Luís Cônsoli, do Departamento de Entomologia e Acarologia da Escola Superior de Agricultura Luiz de Queiroz (Esalq) da USP, em Piracicaba, interior paulista.
O nanoencapsulamento de moléculas de RNAi é o tema do projeto de doutorado do biólogo Cyro von Zuben, aluno do IB-Unicamp orientado por Marques-Souza. Até o fim do ano deverão ser realizados ensaios com nanopartículas de argila, em conjunto com o Instituto de Pesquisas Tecnológicas (IPT), do governo paulista, e com o Laboratório Nacional de Nanotecnologia (LNNano), do Centro Nacional de Pesquisa em Energia e Materiais (CNPEM), para diversas espécies-alvos em estudo no laboratório Blast. Na Austrália, um grupo de pesquisa publicou há pouco tempo um artigo descrevendo o uso de nanopartículas que garantem a integridade e a entrega de RNAi por 30 dias.
Cônsoli também alerta que o processo de seleção de moléculas de RNAi deve considerar o risco de elas afetarem a própria planta ou organismos não alvo da tecnologia, inclusive o homem. De acordo com Marques-Souza, essa é uma preocupação real, mas que pode ser superada. “Quando se seleciona um gene-alvo para atingir o organismo de uma praga, devemos ter o cuidado de não escolher uma sequência gênica que seja complementar a um gene da planta ou de qualquer outro organismo que se alimente dela. Se este cuidado for tomado, o silenciamento gênico só ocorrerá no organismo-alvo”, diz. “A garantia de que o RNAi não terá nenhum efeito em humanos só virá com muito estudo.”
Alexandre Garcia reforça: “A tecnologia é baseada em sequências de RNA. Todos os seres vivos possuem sequências em comum, mas as espécies têm inúmeras sequências únicas que as diferenciam de outras espécies”. Hoje, com a quantidade de informações e bases de dados de genomas à disposição, é possível achar sequências que são exclusivas da espécie-alvo e desenhar moléculas que irão atuar apenas nessas sequências. “Dessa forma, todo o processo se torna específico e exclusivo para o organismo-alvo”, sustenta.
Norma da CTNBIO
Um passo importante para a regulamentação do uso do RNAi tópico no Brasil foi dado no início do ano quando a CTNBio publicou uma normativa definindo a tecnologia como um método que não envolve transgenia e classificando-a de Técnica Inovadora de Melhoramento de Precisão (Timp). Com isso, ela não estará necessariamente sujeita aos mesmos critérios de aprovação exigidos para organismos geneticamente modificados.
“O processo para gerar uma planta transgênica e passar por todos os mecanismos regulatórios globais para aprovar seu cultivo comercial é extremamente caro e demorado, podendo ultrapassar os US$ 100 milhões e 15 anos”, diz Garcia. “O uso da tecnologia de RNAi como produto de aplicação tópica, para o caso do percevejo-da-soja, é mais promissor. Essa tecnologia deverá estar à disposição dos agricultores brasileiros em alguns anos.”
Projeto
RNAi para controle de Tuta absoluta em tomateiro (nº 11/12869-6); Modalidade Auxílio à Pesquisa – Regular; Pesquisador responsável Antônio Vargas de Oliveira Figueira (USP); Investimento R$ 153.800,39.
Artigos científicos
PARTHASARATHY, R. et al. Physiological and cellular responses caused by RNAi-mediated suppression of Snf7 orthologue in western corn rootworm (Diabrotica virgifera virgifera) larvae. PLOS ONE. On-line. 18 jan. 2013.
CAMARGO, R. A. et al. RNA interference as a gene silencing tool to control Tuta absoluta in tomato (Solanum lycopersicum). PeerJ. On-line. 15 dez. 2016.
Autor: Revista Pesquisa Faperj
Fonte: Revista Pesquisa Faperj
Sítio Online da Publicação: Revista Pesquisa Faperj
Data de Publicação: 24/6/2018
Publicação Original: http://revistapesquisa.fapesp.br/2018/06/18/genes-em-silencio/
/i.s3.glbimg.com/v1/AUTH_59edd422c0c84a879bd37670ae4f538a/internal_photos/bs/2018/G/A/LBw7QUQ9qKxh12LYb4pw/161778.jpg)
/s.glbimg.com/jo/g1/f/original/2016/02/27/gemeos2.jpg)




